COVID-19 vaccine still months away for most Americans
Experts say it is much too premature for people to start demanding a shot.
Pfizer and its partner, German company BioNTech, became the first manufacturer to ask the Food and Drug Administration for emergency use authorization for a COVID-19 vaccine Friday, paving the way for some Americans to receive their first doses of the vaccine by the middle to the end of December -- less than one year after the novel coronavirus reached U.S. shores.
The U.S. government's Operation Warp Speed has promised "shots in arms" within 24 hours of an FDA authorization.
But experts say it is much too premature for people to start calling their physicians, demanding a shot.
Although Pfizer has officially asked the FDA for authorization, it will take the agency two to four weeks to review all the data and make a decision. Even then, the government only has enough vaccine doses for about 20 million Americans by the end of this year.
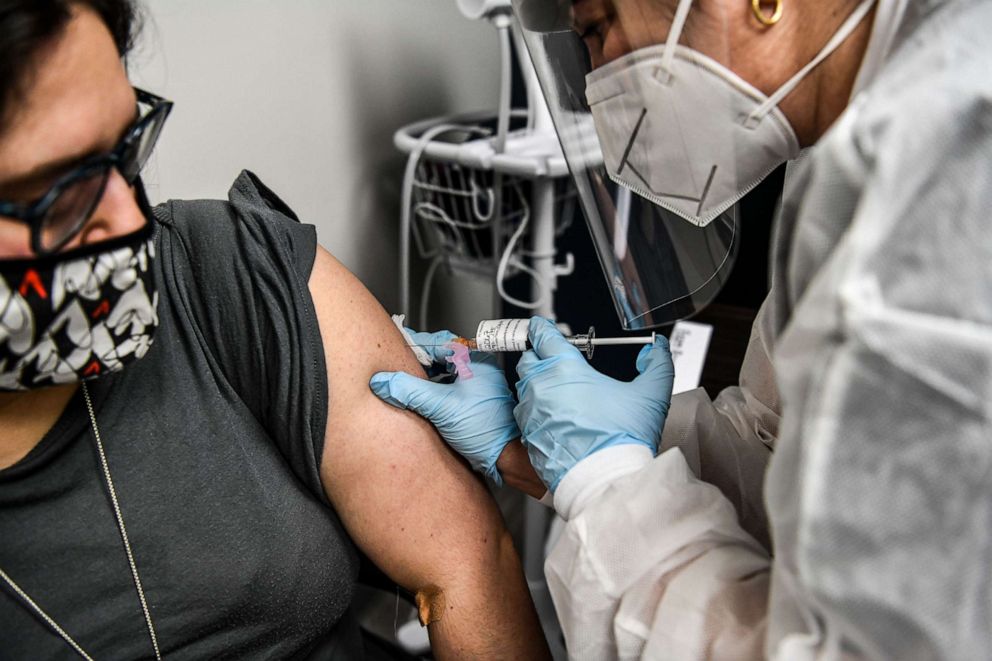
"Not every American will have a needle in their arm immediately following authorization," said Dr. Carlos del Rio, a professor of medicine and global health at Emory University School of Medicine.
"They're going to start with first responders and medical personnel and elderly and people with comorbidities. The regular Joe is probably not going to receive a vaccine until July or August," del Rio predicted.
The FDA's emergency use authorization program, established in 2004, allows the government to permit for limited use vaccines that would be needed in case of a national emergency. Baseline requirements are that there must be an emergency; the potential benefits of the product must exceed the risks; and there must be reasonable belief that "the product may be effective."
The COVID-19 threat qualifies for all of these conditions.
"All of our decisions will continue to be based on good science and the same careful deliberative processes we have always used when reviewing medical products, " FDA Commissioner Dr. Stephen Hahn told the American Medical Association in a video briefing in August.
A critical step in the FDA's authorization process is the review and evaluation of the data pertaining to the safety and efficacy of vaccines by the Vaccines and Related Biological Products Advisory Committee, or VRBPAC, an independent group of experts in infectious diseases and vaccines.
Dr. Paul Offit, director of the Vaccine Education Center and professor of pediatrics in the Division of Infectious Diseases at Children's Hospital of Philadelphia, said, "First the FDA will review all the data, then they will present those data to its advisory committee -- a process that will likely take a few weeks."
The FDA announced Friday that the independent VRBPAC advisory meeting will take place on Dec. 10. The committee members -- comprised of a panel of vaccine experts -- will then recommend whether the FDA should grant limited authorization.
Del Rio believes the FDA's decision to authorize will be "immediate."
The coronavirus vaccine review process will be overseen by the head of the FDA's center for biologics, Dr. Peter Marks, director of the Food and Drug Administration's Center for Biologics Evaluation and Research, who previously vowed to resign if there were outside pressures affecting his organization's review process, Reuters reported.
Marks told Business Insider that the process would be open and transparent, and that it would take "weeks" to review the vaccine applications.
"We have to take the amount of time that we need to take," Marks said. "The caveat here with saying this word 'weeks' is there are situations whereby the quality of what comes in and the complexity of what comes in will determine how long it will take to do a review that we can feel very confident in."
"It should take a couple of weeks for FDA to complete review of thousands of pages of data, go to committees and, if everything checks out as stated in Pfizer press releases, hopefully begin vaccinating in pilot programs in December," Dr. Peter Jay Hotez, dean of the National School of Tropical Medicine, told ABC News.
Pfizer and its competitor, Moderna, just a week behind, have surpassed a key milestone: at least two months of follow-up safety data on the health of the clinical trial participants, a condition it imposed on all vaccine manufacturers in October.
In addition to the FDA, another group of advisers for the Centers for Disease Control and Prevention, the Advisory Committee on Immunization Practices, must also meet. It is comprised of medical and public health experts who make recommendations as to who should first get the vaccines.
Although it is unclear as to when the meeting of the ACIP will take place, Health and Human Services Secretary Alex Azar said on Wednesday that he will seek to move the CDC advisory meeting up, to correspond with that of the FDA independent advisers, so that both groups will issue a final guidance simultaneously.
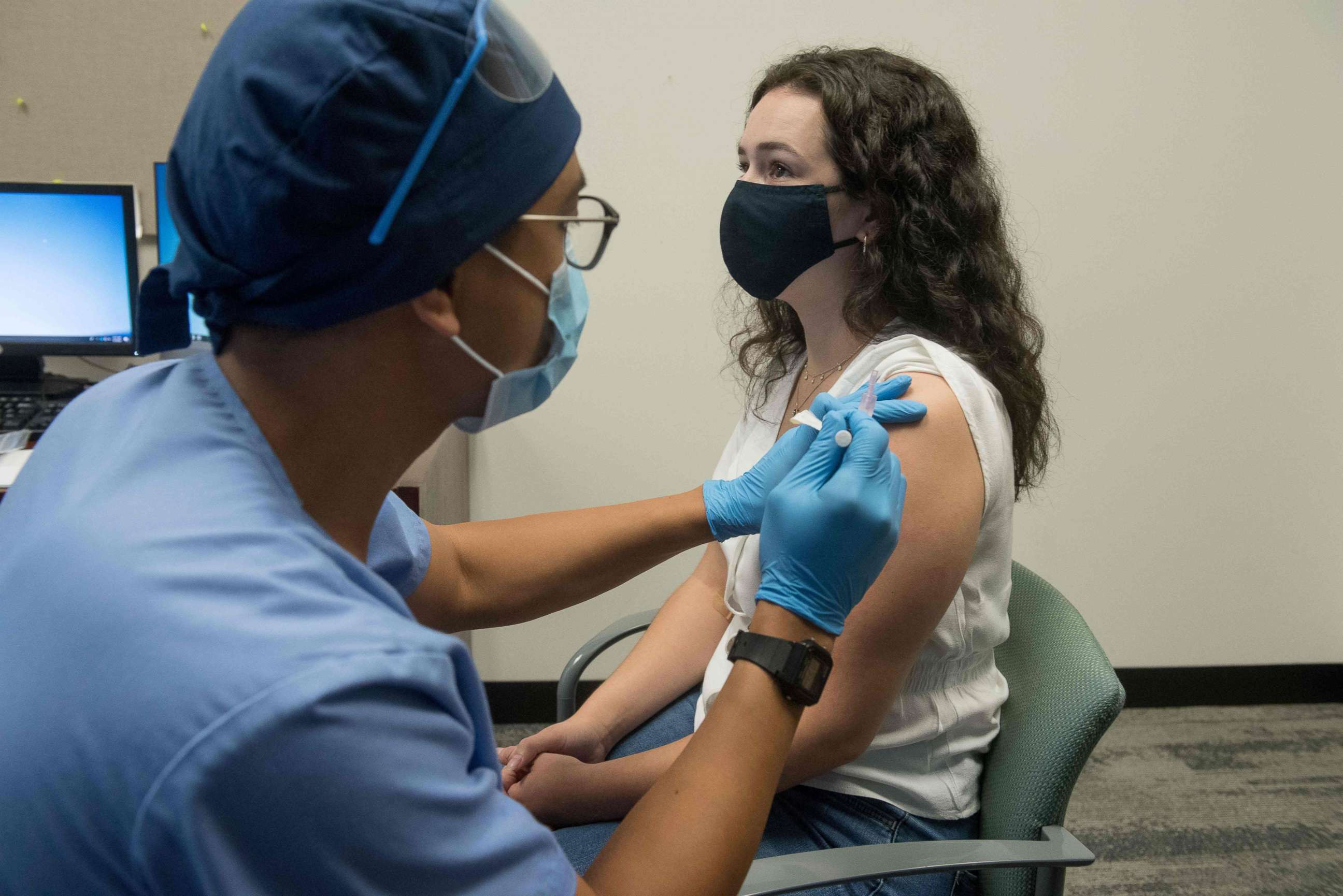
The recommendation of the CDC's committee with respect to who gets the vaccine first may ultimately depend on the analysis of the data from the manufacturers.
With both the Pfizer and Moderna vaccines, Azar said, the U.S. should have access to about 40 million doses by the end of December, allowing 20 million people to get the two-dose regimen.
Despite the positive news, there are major challenges in implementing nationwide inoculations, including "vaccinating 300 million Americans, when 50% of them are skeptical about the vaccine. And logistics are going to be really complicated," del Rio said. "This is not going to be an easy vaccine to roll out."
If all goes according to plan, by next spring "a robust program of vaccination will be underway for the nation, with possibly four vaccines -- two mRNA vaccines, two adenovirus vaccines," and potentially a fifth Novavax particle vaccine, said Hotez.
Related Topics

Popular Reads
ABC News Live